
Sickle Cell Disease
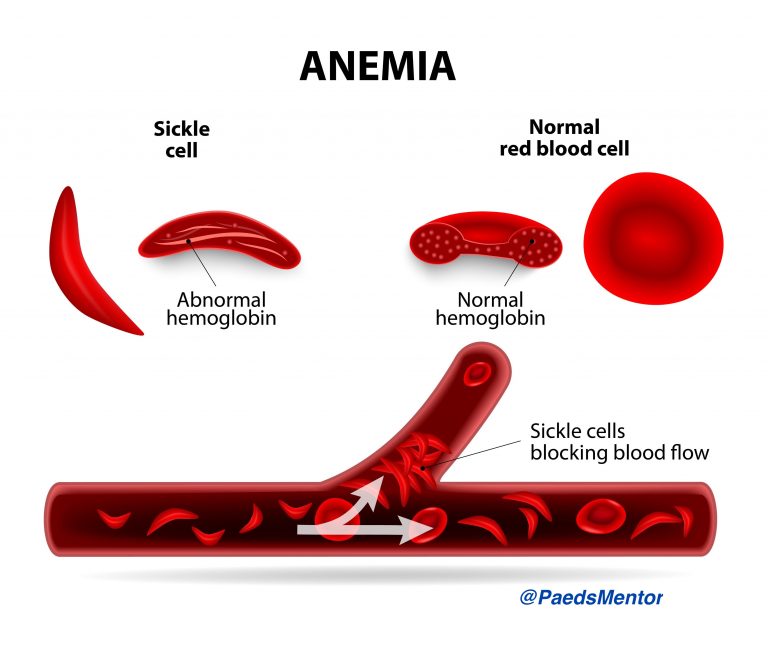
Sickle Cell Disease (SCD) is a group of inherited haemoglobin disorders caused by a mutation in the beta-globin gene (HbS).
This mutation leads to the polymerisation of deoxygenated haemoglobin, which distorts red blood cells into a characteristic sickle shape. These rigid, sticky cells obstruct small blood vessels, leading to chronic haemolysis and recurrent episodes of vaso-occlusive crisis (VOC).
The condition is inherited in an autosomal recessive manner. In the UK, it is most prevalent in individuals of African and Caribbean descent. All babies are offered screening as part of the Newborn Blood Spot (heel prick) Test at 5-8 days of age. Early diagnosis is crucial to initiate prophylactic measures and specialist care, which significantly reduces morbidity and mortality.
Management in a Paediatric Setting
Acute management focuses on the prompt and aggressive treatment of complications. All children with SCD should have open access to a paediatric unit.
1. Painful Vaso-Occlusive Crisis (VOC)
This is the most common reason for hospital admission. The goal is to provide rapid and effective pain relief within 30-60 minutes of presentation.
Assessment: Use an age-appropriate pain score (e.g., Faces Pain Scale, FLACC). Take a detailed history, including the site and intensity of pain, and any analgesia already administered.
Hydration: Oral hydration is preferred, but intravenous fluids are indicated for severe pain, vomiting, or if the child is not settling. Hyperhydration should be avoided due to the risk of fluid overload.
Analgesia: Start with simple analgesia (paracetamol, ibuprofen, or diclofenac) and titrate up to strong opiates as needed. Opiates, such as oral morphine or intranasal diamorphine, are often required for moderate-to-severe pain. For severe, unremitting pain, consider a morphine patient-controlled analgesia (PCA) or nurse-controlled analgesia (NCA), following local protocols.
Non-Pharmacological Measures: Encourage warmth, massage, distraction, and relaxation.
2. Acute Chest Syndrome (ACS)
This is a life-threatening complication characterised by fever, respiratory symptoms, and new infiltrates on chest X-ray. It requires urgent management.
Diagnosis: Be vigilant for chest pain, cough, tachypnoea, or fever in a child with SCD.
Management: Admit for oxygen, IV antibiotics (e.g., cefuroxime plus clarithromycin), analgesia, and IV hydration. Consider a simple or exchange blood transfusion in severe cases or if there is a rapid drop in haemoglobin.
3. Splenic Sequestration Crisis
This is an acute, life-threatening emergency where red blood cells become trapped in the spleen, leading to a sudden, rapid drop in haemoglobin.
Diagnosis: Suspect in any child with SCD who presents with pallor, lethargy, and a rapidly enlarging spleen.
Management: Urgent blood transfusion is the mainstay of treatment.
Recent Developments and Future Directions
Gene Therapies: The most significant development is the approval of gene therapies like exagamglogene autotemcel (Casgevy). This is a one-time curative treatment that uses CRISPR-Cas9 gene-editing technology to increase the production of foetal haemoglobin (HbF). Higher levels of HbF prevent sickling, and trials have shown a significant reduction in VOCs. In the UK, NICE has recently approved this treatment for certain patients with severe forms of SCD.
Novel Drug Therapies:
Crizanlizumab: A monoclonal antibody that targets P-selectin, a molecule involved in cell adhesion. It is used to reduce the frequency of VOCs. However, recent phase III trials have shown no benefit in pain events, leading to its revocation of approval in Europe.
Voxelotor: A small molecule that inhibits HbS polymerisation, increasing haemoglobin’s affinity for oxygen. This drug has been voluntarily withdrawn from the market by its manufacturer due to a lack of clinical benefit.
Rilzabrutinib: A Bruton’s tyrosine kinase (BTK) inhibitor currently under investigation.
Haematopoietic Stem Cell Transplant (HSCT): Allogeneic HSCT remains a potential cure for SCD, particularly with the use of haploidentical (half-matched) donors. Advancements in non-myeloablative conditioning regimens are reducing toxicity and making this a more viable option for a wider range of patients.