
Neurofibromatosis type 1 (von Recklinghaussen disease)
NF1 is an autosomal dominant multi-system disorder of neural crest origin, with an incidence of about 1 in 3,000 live births.
It is caused by a mutation in the NF1 gene on chromosome 17q11.2. Approximately 50% of cases are new, de novo mutations.
The gene encodes for the protein neurofibromin, a negative regulator of the RAS/MAPK signalling pathway. A deficiency of neurofibromin leads to increased RAS activity, promoting uncontrolled cell proliferation, differentiation, and survival. This results in the characteristic lesions of NF1 and an increased risk of malignancy.
Diagnostic Criteria
The diagnostic criteria have been updated to reflect new knowledge, particularly regarding genetics and choroidal abnormalities. A diagnosis is made if a patient meets two or more of the following criteria:
Six or more café-au-lait macules (>5 mm in pre-pubertal or >15 mm in post-pubertal individuals).
Two or more neurofibromas of any type or one plexiform neurofibroma.
Axillary or inguinal freckling.
Optic pathway glioma.
Two or more iris Lisch nodules (iris hamartomas) OR two or more choroidal abnormalities (Lisch nodules are visible on slit lamp, while choroidal abnormalities are visible on fundoscopic exam).
A distinctive bony lesion, such as sphenoid wing dysplasia or bowing of a long bone with or without pseudoarthrosis.
A pathogenic NF1 gene variant identified via molecular testing.
A first-degree relative (parent, sibling, or child) with NF1.
Diagnostic Confirmation and Follow-up:
The diagnosis may not be apparent in early childhood, with approximately 50% of patients meeting the criteria by 1 year of age and 97% by 8 years of age.
Children with only six or more café-au-lait macules should have annual follow-up.
Molecular genetic testing is now a diagnostic criterion and can be very helpful in uncertain cases or for prenatal counselling. It can identify the gene change in up to 95% of individuals.
Consider other differential diagnoses for hyperpigmented lesions, such as McCune-Albright syndrome and Legius syndrome (caused by a SPRED1 gene mutation, which presents with pigmentary findings but not NF1-related tumours).
Clinical Presentation
NF1 is a multi-system disorder with a wide range of manifestations. Regular annual review is crucial for early detection and management of complications.
Skin: Café-au-lait spots, skinfold freckling, cutaneous neurofibromas, subcutaneous neurofibromas, plexiform neurofibromas. The presence of large plexiform neurofibromas or excessive hair growth over a large café-au-lait macule may indicate a plexiform neurofibroma.
Skeletal: Scoliosis (occurs in up to 10% of children, particularly around puberty), bowing of long bones, fractures, and pseudoarthrosis. Sphenoid wing dysplasia can cause pulsating exophthalmos.
Eyes: Optic pathway glioma (OPG), glaucoma, iris Lisch nodules, and choroidal abnormalities. OPGs typically present in early childhood (before 7 years of age).
CNS: Macrocephaly, brainstem and cerebellar gliomas, epilepsy (5-7% of patients), and abnormal vasculature (e.g., moyamoya disease).
Peripheral Nerves: Plexiform neurofibromas can cause peripheral nerve damage or spinal cord compression. There is an increased risk of Malignant Peripheral Nerve Sheath Tumours (MPNSTs), which are rare but life-threatening.
Developmental/Behavioural: Learning difficulties, ADHD, and Autism Spectrum Disorder (ASD) are common. Anxiety, depression, and disordered sleep are also frequently reported.
Cardiac: Pulmonary stenosis, coarctation of the aorta, and mid-aortic stenosis. Hypertension can be caused by renal artery stenosis or phaeochromocytoma.
Endocrine/Other: Precocious or delayed puberty, phaeochromocytoma, and increased risk of other malignancies in adulthood (e.g., breast cancer).
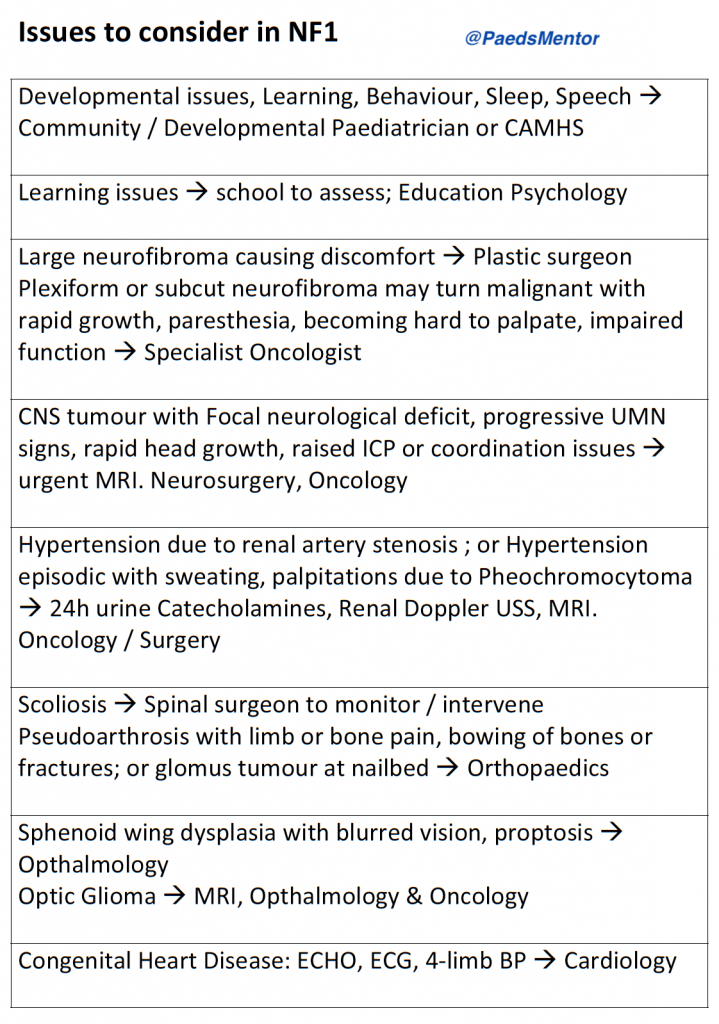
Management and Recent Developments
Management is focused on multidisciplinary care, surveillance, and early intervention.
Annual Monitoring:
Growth (height, weight, and head circumference for under 3s).
Blood pressure measurement.
Developmental surveillance and assessment of learning, behavioural, and emotional issues.
Pubertal assessment.
Ophthalmology review: Annual review with a paediatric ophthalmologist until age 7, then regular checks with an optician.
Physical examination: Monitor skin lesions, examine spine for scoliosis, and check for skeletal changes or pseudoarthrosis. A full neurological examination is essential.
Recent Therapeutic Developments:
Selumetinib (Koselugo®): This is a significant recent development. Selumetinib is an oral MEK inhibitor which targets the overactive RAS/MAPK pathway in NF1. It is approved in the UK for the treatment of symptomatic, inoperable plexiform neurofibromas in paediatric patients aged 3 years and older. Clinical trials have shown it can reduce tumour size and improve associated symptoms like pain, disfigurement, and motor function.
Other MEK inhibitors: Other MEK inhibitors, such as mirdametinib, are in clinical development and may become available in the future.
Ongoing research: Research is ongoing into new treatments, including repurposed drugs and gene-based therapies. The focus is on finding effective treatments for other manifestations of NF1, such as cutaneous neurofibromas and cognitive deficits.
Referrals: Referrals to specialist multidisciplinary teams and services are essential. These may include a clinical geneticist, paediatric neurologist, ophthalmologist, orthopaedic surgeon, oncologist, psychologist, and specialist nurses. UK-based support groups like Nerve Tumours UK and the Childhood Tumour Trust provide excellent resources and support for families.